
发布时间:2024-11-19
近日,美高梅高国华副教授、江南大学杜明亮教授/朱罕研究员、清华大学王定胜教授/庄泽超博士团队合作在《自然·通讯》(Nature Communications)期刊上发表题为“Integrating few-atom layer metal on high-entropy alloys to catalyze nitrate reduction in tandem”的研究论文。
虽然高熵合金(HEA)催化剂因其结构的复杂性似乎有可能打破线性比例关系(LSRs),但多个主成分之间的性能加权平均实际上使其偏离LSRs所施加的对称依赖性具有挑战性。该研究开发了一种“表面熵减”方法,来诱导具有弱亲和力的组分溶出,从而在HEAs表面形成少原子层金属(FL-M)。这些溶出的FL-M超越了传统HEAs的原始构型空间的限制,并与HEA底物协同作用,在复杂反应中作为多个中间体的几何分离活性位点。经FL-M包覆的HEA在电催化还原硝酸盐制氨(NH3)过程中表现出卓越的性能,其法拉第效率高达92.7%,NH3产率为2.45 mmol h-1 mgcat.-1,并具有高长期稳定性(>200 h)。该研究实现了对原子排列的精确控制,从而扩大扩展了已知HEA催化剂所占据的化学空间及其潜在的应用场景。
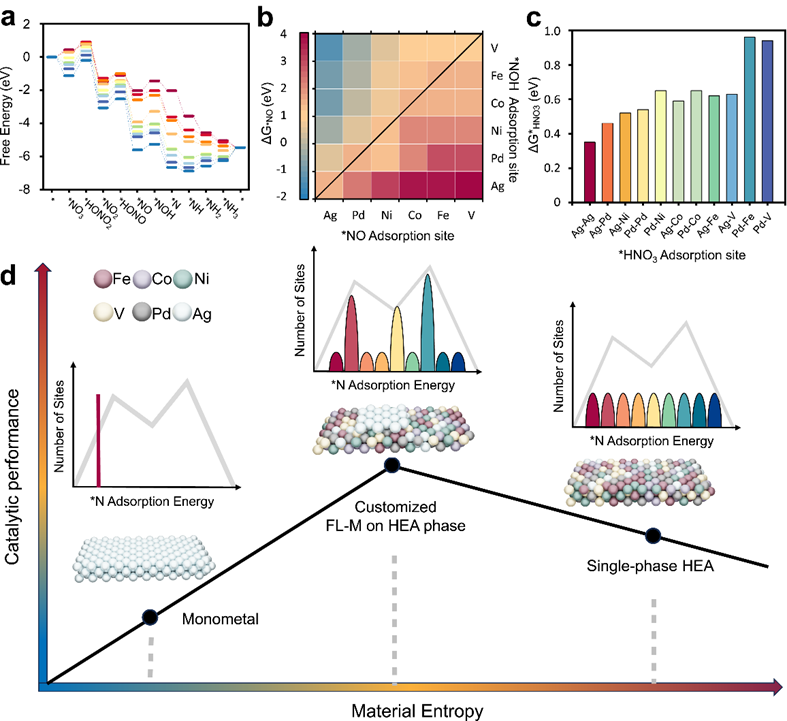
图1.该研究目的。a NitRR在不同单金属上的反应自由能。b HEAhomo中不同金属位点组合下*NO加氢的自由能变化(∆G*NO)。c HEAhomo中不同金属位点上*NO3加氢的自由能变化(∆G*HNO3)。d 随着材料熵的增加,设计的单金属、HEAhomo和定制的FL-Ag/HEA催化剂的电催化活性对比。
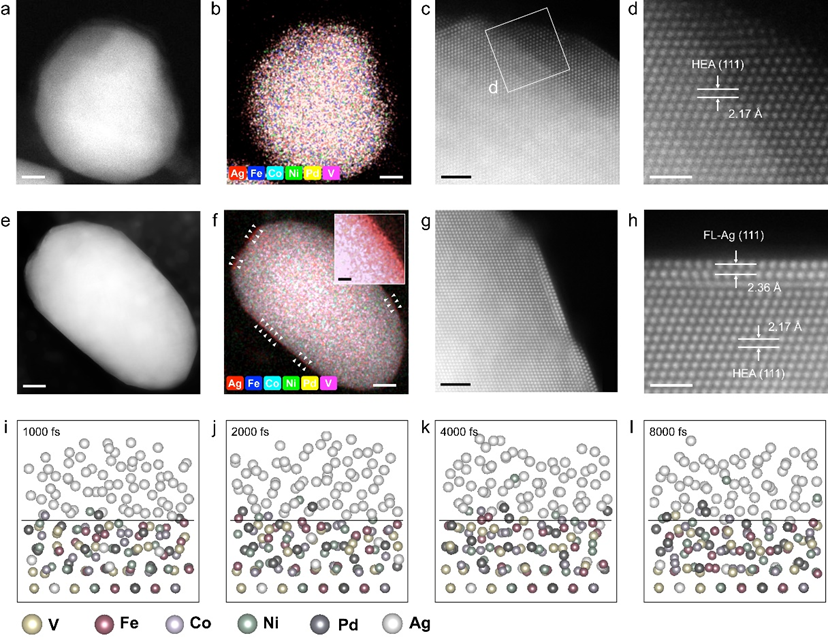
图2. 结构表征。a-d HEAhomo,和e-h 负载FL-Ag/HEA NPs的CNFs的HAADF-STEM图像、STEM-EDS 映射图像和原子分辨率HAADF-STEM图像。i-l MD模拟了1200℃下8 ps内HEA-FL-Ag界面的结构演化。红色虚线是HEA相和FL-Ag的原始分界线,蓝色的圆圈是HEA相向FL-Ag扩散的原子。
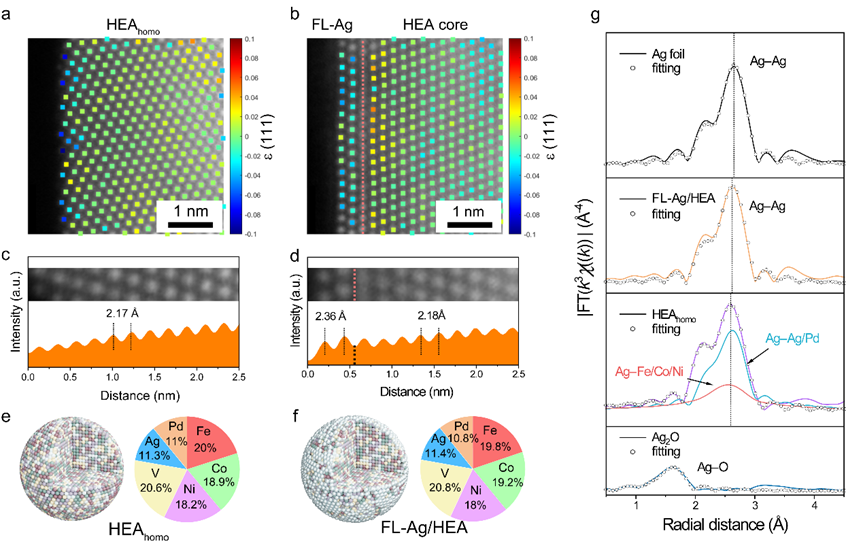
图3. 原子结构分析。利用统计参数估计理论推导出的a HEAhomo和b FL-Ag/HEA中(111)平面的原子分辨应变图。用红色虚线标记纳米界面。c HEAhomo和d FL-Ag/HEA NPs中(111)平面对应的强度分布图。e FL-Ag/HEA和f HEAhomo的三维模型及其组成元素的原子百分比。g FL-Ag/HEA/CNFs、HEAhomo/CNFs、Ag和Ag2O箔的FT-EXAFS和相应的拟合光谱。
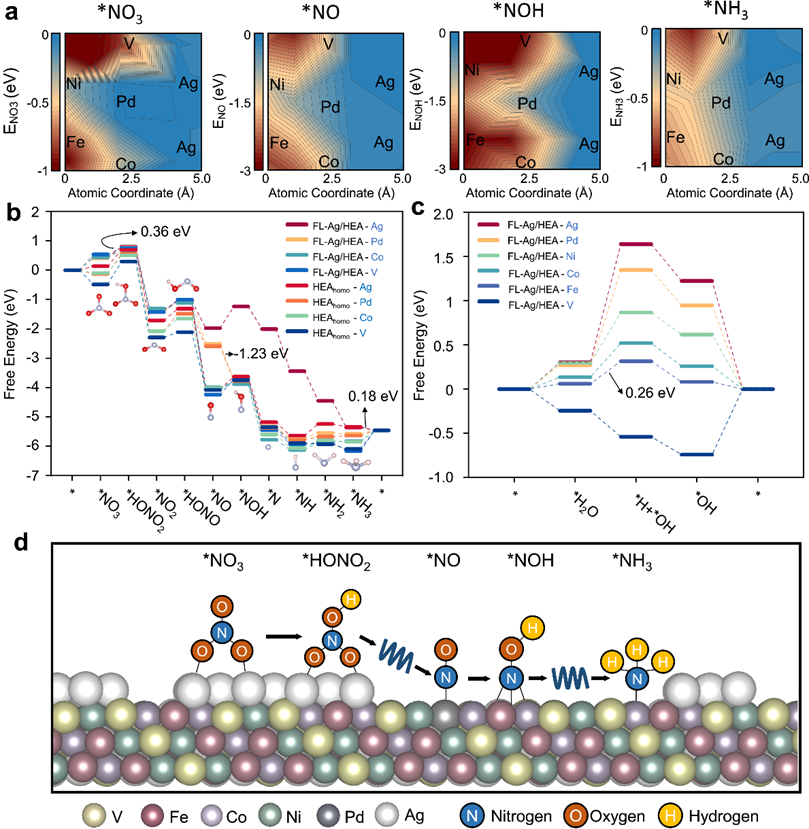
图4. 理论计算。a 中间产物在FL-Ag/HEA表面的吸附能分布(Ei, i = NO3,NO,NOH和NH3)。取吸附能的最低点为0,然后计算其相对值。FL-Ag/HEA和HEAhomo中不同金属位点上的b NitRR和c HER的反应自由能。d FL-Ag/HEA上NitRR的中继催化机理。
高国华副教授、庄泽超博士、王定胜教授、朱罕研究员为论文共同通讯作者,郝嘉策博士与王同德博士为共同第一作者。该研究使用“表面熵减”策略,来设计用于NitRR的FL-Ag/HEA电催化剂。FL-Ag/HEA/CNFs的NH3法拉第效率高达92.7%,NH3产率为2.45 mmol h-1 mgcat.-1,具有显著的长期稳定性(>200 h),优于大多数最先进的电催化剂和传统的Haber-Bosch工艺NH3生产(NH3产率= 0.2 mmol h-1 mgcat.-1)。实验和计算结果突出了FL-Ag在HEA表面对提高NitRR活性的重要性和优势。以HEA和FL-Ag分别作为脱氧和加氢活性中心,可在FL-Ag/HEA上进行中继电催化,其中*NO3在FL-Ag上转化为*NO,而*NO中间体迁移到HEA-FL-Ag界面上邻近的Pd位点,随后由*NO加氢为NH3。该研究得到了国家自然科学基金等资助。
论文链接:https://www.nature.com/articles/s41467-024-53427-7